|

- 现金
- 62111 元
- 精华
- 26
- 帖子
- 30437
- 注册时间
- 2009-10-5
- 最后登录
- 2022-12-28

|
Multi-step regulation of hepatitis B virus entry October 10, 2019
Koichi Watashi, Senior Researcher from the National Institute of Infectious Diseases, Japan, explores the trick for cell invasion in reaching regulation of hepatitis B entryChronic hepatitis B virus (HBV) infection is estimated to affect approximately 257 million people worldwide. For achieving the World Health Organization (WHO) strategy to control hepatitis by 20301, understanding the HBV transmission is of particular importance. Viral transmission is determined by factors derived from both virus itself and host. One of such major factors in host is a cell surface protein that mediates the binding to virus, defined as a viral entry receptor. Sodium taurocholate cotransporting polypeptide (NTCP), a bile acid transporter, has been revealed to function as an HBV entry receptor in 20122. This protein, expressed exclusively in the liver, is an apparent factor determining the HBV infectivity to host.
However, it has been totally unclarified how NTCP mediates HBV entry and determines host susceptibility to HBV infection. In April 2019, Koichi Watashi’s group in National Institute of Infectious Diseases, Japan published a paper in Proc Natl Acad Sci USA that provides a new concept that HBV infection is determined by another molecule that regulates the function of virus-receptor complex, termed as an entry cofactor3.
Cell culture and animal model data so far demonstrate that NTCP is required for HBV infection. However, there is accumulating evidences suggesting that NTCP expression is not sufficient for reproducing HBV infection in cells: HBV infection is not necessarily observed in all the NTCP expressing cells, and the infection in susceptible cells depends on culture condition, irrespective of the NTCP expression level. This data raises the possibility that another factor(s) regulates HBV infection. Koichi Watashi’s group has been pursuing to identify a series of small molecules and bioactive ligands that suppress or facilitate hepatitis virus infection. Their strategy is to take advantage of these agents to probe the mechanisms underlying virus infection.
So far, they have identified a bunch of small molecules that inhibit HBV infection, including cyclosporins, tricyclic polyketides, synthetic retinoids, coumarines, macrolides, thiazolidinediones, and cyclic peptides, which were reported in the context of drug development4. In addition, the group has also found those rather facilitating HBV infection, one of which is epidermal growth factor (EGF). By focusing on this probe, they revealed that EGF receptor (EGFR) regulates HBV infection to NTCP-expressing cells. Even if expressing NTCP, HBV infection was no more observed by deficiency of expression or function of EGFR.
Mechanistically, while NTCP mediates HBV attachment to the cell surface, EGFR was shown to be involved in HBV translocation to the endosomes (Figure). Their data and previous evidences suggest that EGFR interacts with NTCP irrespective of HBV infection: EGFR-NTCP complex is likely to travel between the cell surface and the intracellular vesicles by following the cellular membrane trafficking pathway either in the presence or absence of infection. When HBV attaches to NTCP on the cell surface, it migrates with NTCP-EGFR inside the vesicles through the trafficking. Thus, NTCP functions for the attachment with HBV, and EGFR drives the internalization of HBV-NTCP complex.
Notably, if NTCP does not bind to EGFR, NTCP no longer functions as the entry receptor of HBV, which was shown by the blockade of NTCP-EGFR binding by introducing amino acid mutation and treatment with decoy peptide. This data clearly show that productive infection of HBV requires EGFR as an entry cofactor, in addition to NTCP.
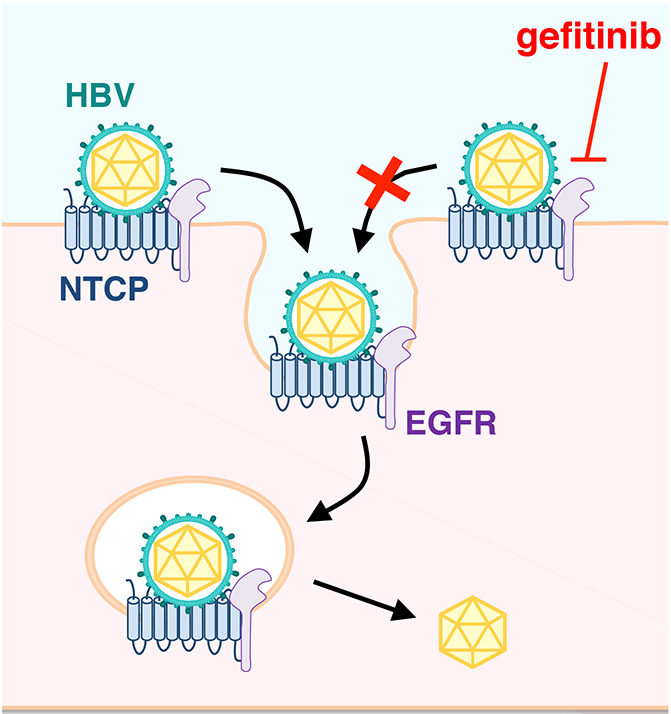 Figure 1. HBV entry. After HBV binds to its receptor, NTCP, HBV-NTCP-EGFR complex is internalised into cells. Gefitinib blocks HBV internalisation through inactivation of EGFRHuman immunodeficiency virus (HIV) uses its receptor, CD4, for cell attachment and requires another involvement of coreceptor, CCR5 or CXCR4 for the following membrane fusion. Hepatitis C virus (HCV) attaches to host cells through CD81 and scavenger receptor class B type I (SR-BI), which then clusters with tight junction proteins, claudin-1 and occludin for internalization. The presenting multi-step regulation of HBV entry is analogous to such examples for the entry mechanism of these viruses. However, it is unique that HBV follows the cellular intrinsic pathway of the membrane protein trafficking that readily occurs in the absence of infection. Figure 1. HBV entry. After HBV binds to its receptor, NTCP, HBV-NTCP-EGFR complex is internalised into cells. Gefitinib blocks HBV internalisation through inactivation of EGFRHuman immunodeficiency virus (HIV) uses its receptor, CD4, for cell attachment and requires another involvement of coreceptor, CCR5 or CXCR4 for the following membrane fusion. Hepatitis C virus (HCV) attaches to host cells through CD81 and scavenger receptor class B type I (SR-BI), which then clusters with tight junction proteins, claudin-1 and occludin for internalization. The presenting multi-step regulation of HBV entry is analogous to such examples for the entry mechanism of these viruses. However, it is unique that HBV follows the cellular intrinsic pathway of the membrane protein trafficking that readily occurs in the absence of infection.
Moreover, as EGFR senses extracellular ligands to change its functionality, HBV susceptibility of cells may be affected by these extracellular ligands, not only by NTCP genetic condition. Next questions include whether only EGFR functions as an entry cofactor of HBV, given that cells express related receptor tyrosine kinases such as fibroblast growth factors, vascular endothelial growth factors, and platelet-derived growth factors. How the entry cofactor drives HBV internalization, and releases HBV afterwards for the productive viral infection? Good news is that this machinery is expected to serve as a target for anti-HBV development. Actually, a peptide that interrupted EGFR-NTCP binding blocked HBV internalization and infection.
Similarly, EGFR tyrosine kinase inhibitor, gefitinib, a clinically used anti-cancer drug, inhibited HBV infection. Further analysis of targeting this machinery for developing a new anti-HBV strategy is of great demand in the near future. Albeit still many blackbox, we have had a significant gain of understanding in this decade on the mechanism underlying HBV infection and determining the susceptibility to HBV. Further analysis on HBV infection is expected to develop new strategies to control the worldwide hepatitis.
|
|




 发表于 2019-10-11 13:38
发表于 2019-10-11 13:38