|
  
- 现金
- 3700 元
- 精华
- 16
- 帖子
- 1790
- 注册时间
- 2002-12-9
- 最后登录
- 2021-4-14




|
6楼
 发表于 2005-4-2 06:13
发表于 2005-4-2 06:13
Resistance
Entecavir has demonstrated activity against HBV replication in vitro with IC50 values ranging from 0.36 nM to 3.6 nM. The active intracellular moiety of ETV, ETV-triphosphate, is a competitive inhibitor of dGTP and functions as a non-obligate chain terminator. Cell culture studies have shown that viruses with the LVD resistance-associated amino acid substitutions rtM204V/I and rtL180M in the HBV polymerase display cross-resistance to ETV, having approximately 5- to 30-fold reduced susceptibility in vitro. Resistance analyses from early Phase II studies provided evidence that substitutions at positions rtT184, rtS202, and/or rtM250 are associated with ETV resistance, but developed only when LVD resistance mutations were present. The addition of substitutions at rtT184, rtS202, and/or rtM250 together with the LVD-resistance mutations, rtL180M and rtM204V, in recombinant viruses resulted in 38- to 2,000-fold reduced susceptibility to ETV in vitro.
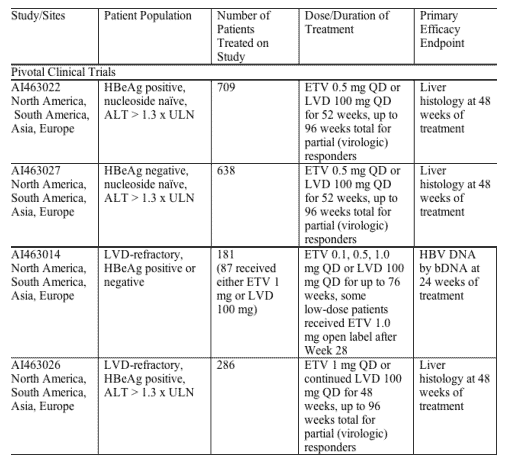
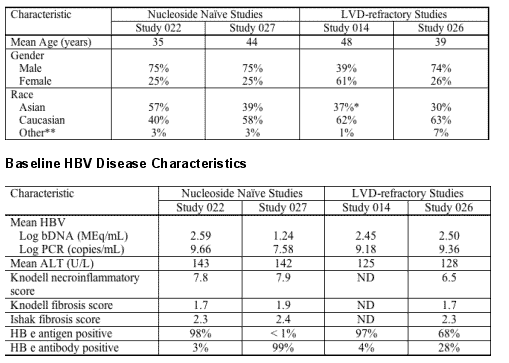
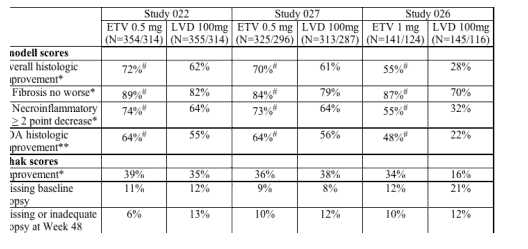
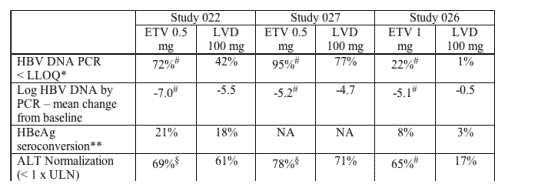
The efficacy of ETV was examined in both nucleoside-naive and LVD-refractory patient populations. In nucleoside-naive Studies 022 (HBeAg positive subjects) and 027 (HBeAg negative subjects), 83% (541/653) of patients on 0.5 mg QD ETV treatment were suppressed with HBV DNA < 400 copies/mL as quantified by the COBAS Amplicor HBV Monitor PCR assay at week 48 compared to 59% (363/619) of patients on 100 mg QD LVD treatment. Genotypic and phenotypic analyses of paired clinical isolates obtained at study entry and Week 48 were performed to monitor baseline and emerging amino acid substitutions and to determine their impact on virologic response to ETV. In these studies, no ETV-associated resistant substitutions (T184S/A/I, S202G, M250L) emerged in any isolate on ETV therapy by 48 weeks. Two subjects experienced virologic rebound on ETV treatment but had no detectable amino acid changes emerge on treatment and no change in phenotypic susceptibility to ETV, ADV or LVD.
Studies 014 and 026 examined the efficacy of ETV 1 mg QD compared to LVD 100 mg QD in patients with LVD-refractory HBV with prior LVD experience. In these studies, LVD-resistant substitutions rtL180M and rtM204V/I were detected in > 80% of baseline isolates from both the ETV and LVD arms and these substitutions were maintained during the study, presumably because of the selective advantage in the presence of LVD and ETV. In Studies 014 and 026, 21% (36/174) of patients on ETV were suppressed to below 400 copies/mL HBV DNA by PCR assay at week 48 compared to 1% of patients on LVD.
Genotypic analyses determined that LVD-resistance substitutions L80V, L180M, M204V or I emerged in the HBV of 17% (7/42) of patients on ETV by Week 48 in Study 014. These substitutions often arose in the context of mixtures at these sites at baseline and other LVD-resistance mutations at baseline. Despite the emergence of LVD-resistance substitutions, the viral load in 4 of 7 patients was suppressed below 300 copies/mL (LLOQ) and the other 3 subjects experienced > 2 log10 reductions in viral load at the time the isolate developed the LVD-resistant mutations. ETV-associated resistance substitutions at T184 developed on 1 mg ETV therapy in 5 (12%) patients after week 48 in Study 014 and coincided with rebounds in viral load. In Study 026, substitutions at RT residues rtI169, rtT184, rtS202 and/or rtM250 emerged on therapy in 9% (12/134) of ETV subjects with Week 48 data. In all cases, the ETV-resistant substitutions emerged when pre-existing LVD-resistant changes were present.
The supportive Study 015 examined the antiviral activity of open label ETV 1 mg QD in 9 orthotopic liver transplant (OLT) recipients who were > 100 days post-transplant and had recurrent HBV infection despite prophylaxis with anti-HBV agents. In this study, virologic rebound occurred in 6 out of 8 patients - one in the first year therapy, one in the second year, and four in the third year while 2 patients maintained HBV DNA suppression with no rebound out to 127 and 131 weeks of therapy. Seven of the eight patients showed the development of ETV-resistance substitutions at S202G or I (n=5), T184S/I/A/L/F (n=4) or M250V (n=1), and these substitutions were linked to LVD-resistant changes L180M and M204V.
The substitutions at rtI169, rtT184, rtS202 and/or rtM250 were associated with phenotypic ETV resistance. The median fold change from reference of ETV susceptibility was 38 (range 12-2139) for the ETV failure isolates (> 400 copies/mL HBV DNA) that developed ETV-resistance substitutions at 48 weeks in Study 026 (n = 15) and 83 (range 12-10022) for all ETV failure isolates from Studies 026 and 015 > 48 weeks (n = 22).
Overall, the following conclusions can be summarized from the resistance data collected in Studies 022, 027, 014, 026, and 015.
* Greater proportions of nucleoside-naive subjects (83%) with chronic HBV infection achieved HBV DNA levels < 400 copies/mL on ETV treatment compared to LVD-refractory subjects (21%).
* Genotypic or phenotypic evidence of resistance to ETV in nucleoside-naive patients chronically infected with HBV (n = 430) has not been observed up to 48 weeks of 0.5 mg QD ETV treatment in Studies 022 and 027, including 2 subjects in Study 022 who experienced a confirmed virologic rebound.
-7.4% (14/190) of LVD-refractory subjects treated with 1 mg ETV had evidence of emerging ETV-resistance substitutions by week 48.
-ETV-associated resistance substitutions at rtI169, rtT184, rtS202, and/or rtM250 emerged concomitant with LVDÐresistant mutations at rtL180 and/or rtM204 and were associated with virologic rebound upon prolonged ETV therapy.
-Overall, 4 ETV treated subjects exhibited a confirmed rebound in their HBV DNA levels of > 1 log10 by week 48:
2 isolates from Study 022 with no evidence of ETV-resistant substitutions or other genotypic changes
One isolate from Study 015 that developed an rtT184A
One isolate from Study 026 that developed an rtT184A/S
-LVD-resistance substitutions L80V, L180M, M204V or I can emerge in the HBV of patients on 1 mg ETV by Week 48. These substitutions often arise in the context of mixtures at these sites at baseline and other LVD-resistant mutations at baseline.
-Even when LVD-resistant mutations emerged in HBV on ETV therapy, ETV can suppress HBV DNA levels to below detection limits.
-> 2 log10 reductions in viral load and viral load suppression below 400 copies/mL HBV DNA can occur in subjects with LVD-resistance in their HBV at baseline when treated with 1 mg ETV.
-Cross-resistance to ETV was not observed with ADV-resistant HBV.
-HBV developing ETV resistance-associated substitutions in addition to LVD-associated resistance substitutions remained resistant to LVD.
Studies evaluating treatment responses to ETV and monitoring resistance to ETV beyond 48 weeks of dosing are ongoing.
Summary of Non-clinical Toxicology and Carcinogenicity
Overview of General Non-clinical Findings
Entecavir is efficiently phosphorylated to ETV-triphosphate (TP) by cellular nucleoside kinases. By competing directly with the natural deoxyguanosine triphosphate (dGTP), ETV-TP potently inhibits each of the 3 distinct activities of the HBV viral polymerase: priming, reverse transcription of first-strand DNA synthesis, and the DNA-dependent DNA polymerase activity responsible for second-strand DNA synthesis. Entecavir has little activity against the DNA polymerase of mitochondria.
The pharmacokinetic (PK) characteristics of entecavir in mice, rats, rabbits, dogs, and monkeys are comparable to those in humans indicating the acceptability of these species for the toxicological assessment of ETV.
Species-specific, reversible CNS inflammation was seen in dogs administered doses that achieve ~51 times the exposure to ETV in humans at clinically proposed doses. It was concluded that this is not relevant to human safety. Other target organs in repeat-dose studies in animals were the kidneys, liver, lungs, skeletal muscle and testis. Data from a 1-year study in monkeys indicated that there was no target organ toxicity in monkeys at exposures to ETV ~136 times those in humans.
Long-term dosing of ETV was evaluated in a woodchuck model of chronic HBV. In this study, woodchucks received a daily dose of ETV equivalent to the 1 mg human dose for 2 months and then were maintained with weekly dosing for up to 3 years. Viral suppression was maintained through 3 years of treatment with no evidence of emergence of resistant HBV. The applicant reported survival rates of 40% and 80% for animals treated for 14 and 36 months, respectively, compared to a survival of 4% in historical controls. Of most interest, the occurrence of hepatocellular carcinoma was significantly reduced in the animals treated long-term compared to historical control animals.
Overview of Carcinogenicity Studies
In a battery of genetic toxicology studies, ETV was an in vitro mutagen in mouse lymphocytes and clastogenic in vitro in human lymphocytes (without metabolic activation). However, ETV was negative in an Ames assay as well as a mammalian-cell gene mutation assay and a cell transformation assay. It was also negative in two in vivo assays, one for the induction of micronuclei and one for the induction of unscheduled DNA synthesis in primary liver cells.
Carcinogenicity studies in Sprague Dawley rats and CD-1 mice were conducted. Increased incidences of tumors were observed in both the studies. The results of these studies were presented to the Executive Carcinogenicity Assessment Committee (ECAC) on June 17, 2003. The key results of these studies are presented in tabular format in Appendix B. The outcomes of the two studies were as follows:
Rat Carcinogenicity Study: The oncogenicity potential of ETV was investigated in male rats at oral gavage dosages of 0.003 (low), 0.02 (mid), 0.2 (high) or 1.4 mg/kg/day (highest) and in females at dose levels of 0.01 (low), 0.06 (mid), 0.4 (high) or 2.6 mg/kg/day (highest) in comparison with untreated controls for a period of 104 weeks.
The no observed effect level (NOEL) for neoplasia was 0.2 mg/kg/day for males and 0.06 mg/kg/day for females. At tumorigenic doses, systemic exposures were 35- and 4-times that in humans (1 mg daily dose) in male and female rats, respectively.
Treatment-Associated Tumors:
1. Hepatocellular adenomas in female rats were significant (p=0.005) at the highest dose level. Combined adenomas and carcinomas in the female rats were also significant (p=0.005) at the highest dose. In female rats, the combined incidence of adenomas and carcinomas was 1% (controls), 4% (low), 5% (mid), 2% (high) and 18% (highest).
2. Brain gliomas were significant (p=0.025) at the highest dose in both male and female rats. In male rats, the incidence was 0% (controls), 2% (low), 2% (mid), 3% (high) and 7% (highest). In female rats, the incidence was 0% (controls), 0% (low), 2% (mid), 0% (high) and 5% (highest).
3. The skin fibromas in female rats were significant (p=0.025) at the high and highest doses. In female rats, the incidence was 0% (controls), 0% (low), 2% (mid), 3% (high) and 5% (highest).
Mouse Carcinogenicity Study: The oncogenicity potential of ETV was investigated in mice at oral gavage dosages of 0.004 (low), 0.04 (mid), 0.4 (high) or 4.0 mg/kg/day (highest) in comparison with untreated controls for a period of 104 weeks.
The NOEL for neoplasia was 0.004 mg/kg/day for males, based on pulmonary adenomas; for all other tumors in males and females, the NOEL was 0.4 mg/kg/day. At the tumorigenic dose in male mice, systemic exposure was 3-times that in humans (1 mg daily dose).
Treatment-Associated Tumors:
1. Lung adenomas were significant (p=0.005) in male mice (mid, high and highest) and in the female mice at the highest dose (p=0.005); lung carcinomas in both male and female mice were significant (p=0.005) at the highest dose. Combined lung adenomas and carcinomas were significant (p=0.005) in male mice at the mid, high and highest dose levels and in the female at the highest dose level (p=0.005. In male mice, the combined incidence of adenomas and carcinomas was 12% (controls), 20% (low), 26% (mid), 40% (high) and 58% (highest). In female mice, the combined incidence of adenomas and carcinomas was 20% (controls), 13% (low), 10% (mid), 35% (high) and 52% (highest).
2. Hepatocellular carcinomas in male mice were significant (p=0.005) at the highest dose level. Combined liver adenomas and carcinomas were also significant (p=0.005) at the highest dose level in the male mice. In male mice, the combined incidence of adenomas and carcinomas was 11% (controls), 9% (low), 8% (mid), 16% (high) and 25% (highest).
3. Vascular tumors in female mice (hemangiomas of ovaries and uterus and hemangiomas/hemangiosarcomas of spleen) were significant (p=0.005) at the highest dose level. In female mice, the incidence of vascular tumors was 16% (controls), 23% (low), 29% (mid), 26% (high) and 64% (highest).
The ECAC found that the carcinogenicity studies in mice and rats were adequately designed and conducted. The committee judged the results of ETV carcinogenicity studies. They concluded that ETV was a carcinogen in rodents. The committee concluded that ETV produced tumors in both species and both genders, and these results suggest a potential cancer hazard to patients.
At the request of the sponsor, the results of the carcinogenicity studies were presented to the full FDA CAC (CAC), a committee that has been designated as the arbiter of disputes between applicants and review divisions regarding the relevance of results in carcinogenicity studies. The CAC met with the applicant and the Review Team on January 7, 2005 and concluded that hepatocellular adenomas and carcinomas in female rats, skin fibromas in female rats and brain gliomas in both male and female rats were relevant. The committee also agreed that in the mouse carcinogenicity study, liver tumors in males and vascular tumors in females as well as lung tumors in both sexes were relevant to human safety evaluation.
Common Adverse Events, Serious Adverse Events, and Laboratory Abnormalities
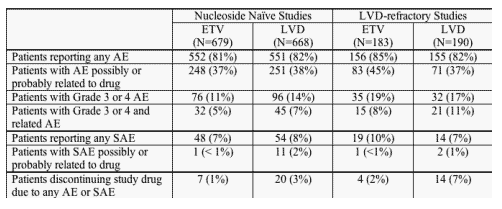
FDA review of the safety database for the pivotal trials confirmed the applicant's analysis of common adverse events (AEs), serious adverse events (SAEs), and laboratory abnormalities. All patients who received at least one dose of blinded study medication in the pivotal trials were included in the safety analyses. The review included assessment of proportions of patients who experienced AEs and SAEs according to severity, relationship to blinded study drug, and action required to manage the event (interruption or discontinuation of study drug). The safety review evaluated clinical events and laboratory abnormalities according to assigned treatment and over 2 study periods (on blinded treatment and off treatment). Organ systems identified as potential targets in the non-clinical animal studies were specifically reviewed. The review evaluated safety in each of the studies individually and also pooled the analyses of nucleoside-naive patients (Studies 022 and 027) and LVD-refractory patients (Study 014 groups receiving ETV 1 mg or LVD and Study 026). Summary results of the pooled analysis will be presented below. Minor differences between the applicant's results and the FDA's results can be attributed to slightly different methods of defining visit windows and conducting the analyses and do not alter the final conclusions.
AEs were reported frequently in the nucleoside-naive patients although there were few differences in the pattern of AEs reported by ETV-treated patients compared to LVD-treated patients. On treatment AEs reported in > 5% of patients in either arm in the nucleoside-naive studies included: headache, upper respiratory infection, nasopharyngitis, cough, pyrexia, abdominal pain, diarrhea, fatigue, arthralgia, dizziness, nausea, influenza, sore throat, rhinorrhea, dyspepsia, increased ALT, increased blood amylase, back pain, and myalgia. Most of the reported events were mild and not considered related to study treatment. The proportions of patients with reported AEs considered by the investigators to be possibly or probably related to blinded study drug were similar in the 2 treatment groups (ETV 37%, LVD 38%). AEs were reported in smaller numbers of nucleoside-naive patients during the off-treatment follow-up period and very few events were observed in > 5% of patients (increased ALT in ETV 3% and LVD 11%, headache in ETV 5% and LVD 6%). During the off-treatment period, only increased ALT occurred more frequently in LVD patients than ETV patients. Other AEs occurred in similar numbers of patients in both groups.
The pattern of commonly reported AEs was very similar in the LVD-refractory patients. On treatment AEs reported in > 5% of patients in either arm in the LVD-refractory studies included: headache, upper respiratory infection, abdominal pain, fatigue, cough, nasopharyngitis, pyrexia, diarrhea, arthralgia, dizziness, nausea, sore throat, dyspepsia, ALT increased, back pain, and myalgia. Most of the events were described as mild and not related to study drug. In this population, increased ALT was reported more frequently in patients receiving LVD (11%) than in those receiving ETV (3%) and fever and sore throat were reported more frequently in ETV patients (9% and 7%, respectively) than in LVD patients (4% and 2%). Reflective of the relatively small proportion of LVD-refractory patients who entered off-treatment follow-up, few patients experienced AEs during the off-treatment period. There were no significant differences in the pattern of off-treatment AEs between the treatment groups.
The number of patients who developed SAEs (death, hospitalization, cancer, congenital anomaly, life-threatening condition, or other medically significant event) while on study was small. Similarly, the number of patients discontinuing their assigned study drug because of an AE or SAE was low, 1% for ETV-treated patients and 4% for LVD-treated patients.
|
|